INTRODUCTION
The Bacillus licheniformis YdaP gene encodes for pyruvate oxidase (POX) (EC: 1.2.3.3) which is a peripheral membrane associated flavoprotein dehydrogenase that belongs to the thiamine diphosphate-dependent enzymes (Cronan, 1995). The thiamine diphosphate-dependent enzymes catalyze the oxidative decarboxylation of pyruvate to acetate and CO2 (Lako et al., 2018). These enzymes are present in a variety of microorganisms from diverse ecosystems including Escherichia coli (Mather et al., 1982), Corynebacterium glutamicum (Schreiner and Eikmanns, 2005), Staphylococcus aureus (Patton et al., 2001; Zhang et al., 2017), Aerococcus viridans (Juan et al., 2007), and Lactobacillus plantarum (Sedewitz et al., 1984; Goffin et al., 2006; Lorquet et al., 2004). These enzymes have been widely studied due to their importance in biotechnological applications (Tomar et al., 2003) and have been well characterized (Lako et al., 2018). The interest in the POX (Pox; 1.2.3.3) is fueled by its potential capacity to produce important commodity chemicals including acetate in the presence of oxygen and inorganic phosphate. This enzyme requires thiamine diphosphate (ThDP), Flavin adenine diphosphate (FAD), and Mg2+ cofactors for its function in catalyzing the oxidative decarboxylation of pyruvate generating acetate (Tittmann et al., 2005). Characterization of the YdaP enzyme has revealed that it is typically composed of four identical subunits in their native state, with each subunit containing one molecule of the Mg2+ cofactor and ThDP. The subunit of this enzyme forms a lose dimer with ThDP and tight homotetramer in the presence of FAD. The enzyme has been expressed and purified and shown to have a molecular weight of 252 KDa (Lako et al., 2018), with seeming stability in purified form in the presence of ThDP. Several structured studies have been performed with the goal of defining the molecular basis of the functions of the family of these proteins (Johnson and Overigton, 1993; Muller et al., 1994; Juan et al., 2007). Most revealing were the two reports describing crystal structures of the POX (L. plantarum, PDB: accession number 2EZ9; Aerococcus viridian, and PDB: accession number IV5E). Comparative analysis of these structures reveals that the overall fold is conserved (Juan et al., 2007). The closest structural homologue was identified as POX, PDB: 2EZ9 from L. plantarum, which shared 35% sequence identity to YdaP (Z-score >6.0) [Figure 1]. As a result PDB: 2EZ9 was selected as a template for the modeling of B. licheniformis YdaP protein. This enzyme represents one of the first moderately thermophilic Bacillus 9A that expresses a highly active, thermostable protein. YdaP enzyme exhibited a very wide range of substrate specificity. The gene encoding this enzyme has been cloned, expressed, purified, and characterized extensively (Lako et al., 2018). Furthermore, the structure shows extensive interactions in the subunit-subunit interface which is significantly different from the other group of POX and might be responsible for the variation in biochemical properties between the species (Muller et al., 1994). In this study, the three-dimensional structures of the POX: 2EZ9 and YdaP were generated using the homology modeling techniques (Sutcliffe et al., 1987; Martin et al., 1994; Muller et al., 1994; Sănchez and Săli, 1997) to compare the quaternary structures of these bacterial POXs with respect to the enzyme substrate interaction and subunit-subunits interface that might be related to the different biochemical characteristics (Bowie and Eisenberg, 1991).

Figure 1: A secondary structure alignment of Bacillus licheniformis YdaP and Lactobacillus plantarum pyruvate oxidase (1 poxa). The alignment was used to generate the B. licheniformis YdaP 3D model
MATERIALS AND METHODS
Modeling of the YdaP Enzyme B. licheniformis and POX of L. plantarum
The sequence alignment of the deduced amino acids of B. licheniformis YdaP and the L. plantarum POX (GenBank: B. licheniformis, accession number: CBE70488 and L. plantarum, accession number: P 37063, respectively) [Figure 1] were used for model building using the MODELLER 9v4 program (Blundell et al., 1987; Šali and Blundell, 1993; Šali and Overigton, 1994; ; Šali et al., 1995) The closest structural homologue was identified as PDB: 2EZ9 from L. plantarum, which shared 35% sequence identity to YdaP (Z-score > 6.0) (Figure 2A). As a result PDB: 2EZ9 was selected as a template for the modelling of B. licheniformis YdaP protein [Figure 2B]. A superimposition of the structural model of B. licheniformis YdaP with L. plantarum POX shows overall structural similarity [Figure 2c]. YdaP shares 35% sequence identity with L. plantarum. The assessment and validation of the model stereochemistry were carried out using the RAMPAGE (Lovell et al., 2001) software. This program analyses and plots Ψ and Ø angles in the structure. The Ψ, Ø plot for the model structure is considered as a reliable method of evaluating torsion angles and has become an important strategy for validation of protein model structures (Kleywegt and Jones, 1998). Overall Ψ, Ø distribution in B. licheniformis YdaP structure was shown to be good [Figure 3].
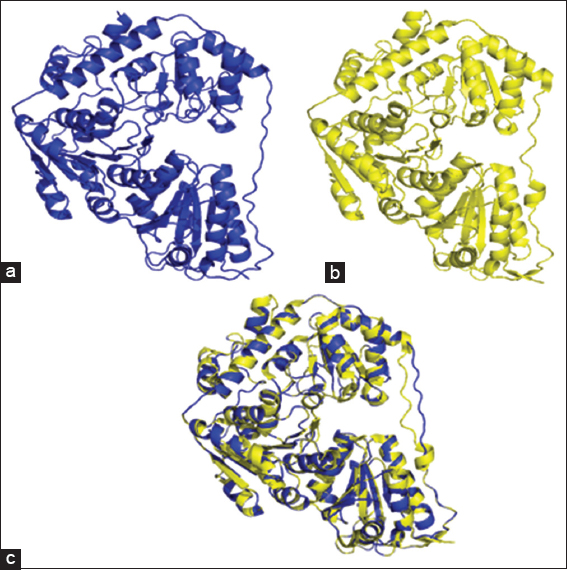
Figure 2: (a) Cartoon representation of the 3D crystal structure of the Lactobacillus plantarum pyruvate oxidase subunit; (b) homology structure model of Bacillus licheniformis YdaP subunit; (c) superimposed structural models of L. plantarum POX and B. licheniformis YdaP 3D
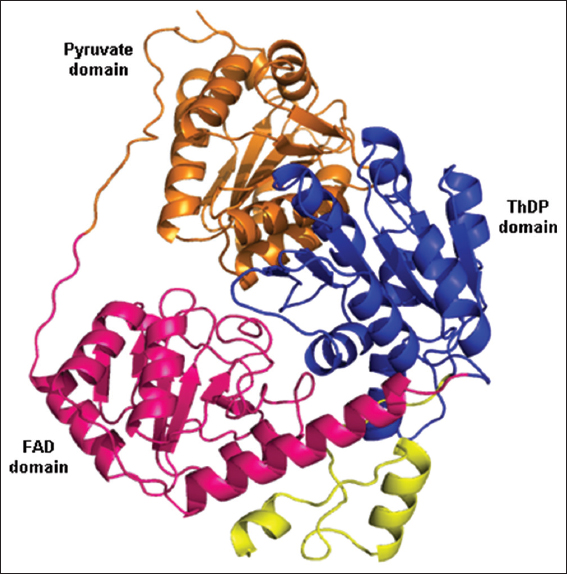
Figure 3: Cartoon representation of the YdaP subunit
RESULTS AND DISCUSSION
Description of the Model Structure
The B. licheniformis YdaP model suggested that the protein monomer was comprised three distinct domains, separated by loops [Figure 4]. These domains were identified using homology search of the secondary structure alignment between B. licheniformis YdaP and L. plantarum POX. The entire globular structure consisted of 22 α-helices and 21 β-sheets. The N-terminal domain was commenced with a long core domain stretching from residues 3–183 (in orange). This was followed by the FAD domain, which spans residues 184–351 (in pink). These domains lead to the long ThDP domain, stretching between residues 352 and 550 (in blue). The C-terminal primary structure consists between residues 550 and 572 (in yellow) was thought to be a membrane anchor (Neumann et al., 2008). These features are found in all elucidated POX structures (Muller and Schulz, 1993; Neumann et al., 2008; Wille et al., 2006).
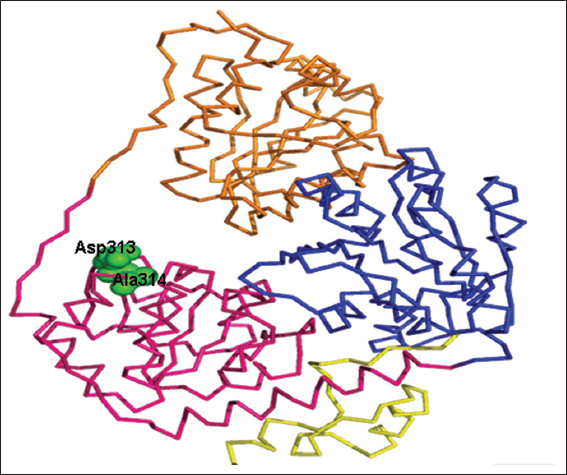
Figure 4: Ribbon representation of residues Asp313 and 314 (in green) predicted to be involved in binding to the flavin adenine diphosphate cofactor
The cofactor FAD was predicted to bind to the YdaP enzyme at Asp313 and Ala314 in the FAD domain [Figure 5], which corresponded to residues Asp323 and Ala324 in L. plantarum POX and appeared to be conserved in both structures. This prediction was based on the fact that these residues were highly conserved in POXs from different organisms (Wille et al., 2006).
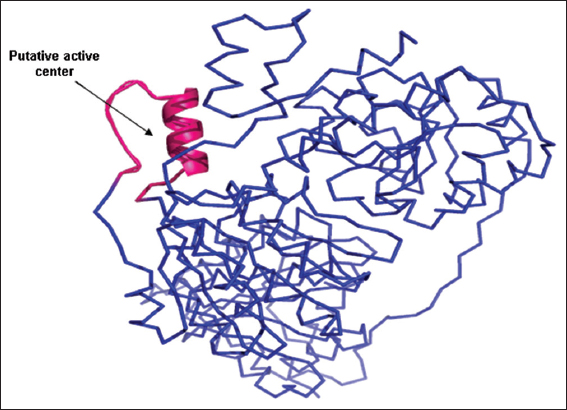
Figure 5: Proposed active center of Bacillus licheniformis YdaP
Topological Description of the YdaP Model
The model structure of YdaP displayed an overall topology similar to the experimentally determined structures of POX from L. plantarum (Bajorath et al., 1994; Wille et al., 2006), E. coli, and A. viridans (Juan et al., 2007). The active site structure showed considerable topological homology in both models and template structures (Sánchez and Šali, 1997; Shi et al., 2001). However, the YdaP substrate-binding pocket was similar to the equivalent site in L. plantarum POX, supporting experimental data showing that YdaP accepted pyruvate and some larger branched chain substrates (Lako et al., 2018). The ThDP motifs of both model structures exhibited a similar structure, their sequences homology of 52%. This observation suggests that the two enzymes are closely related and could indicate an evolutionary relationship (Arnold, 1998) [Figure 6].
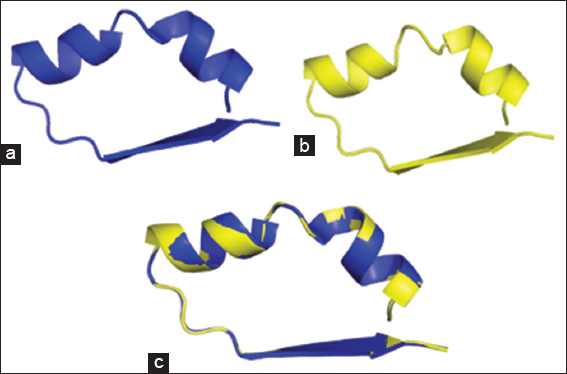
Figure 6: Cartoon representation of (a) ThDP motif of YdaP from Bacillus licheniformis; (b) ThDP motif of LpPOX from Lactobacillus plantarum; (c) superimposed ThDP motifs of both structure models
Disulfide Bridges
A detailed analysis of the overall model structure of B. licheniformis YdaP performed using the SSprov 4.5 SCRATCH protein predictor (/www.ics.uci.edu/~baldig/scratch/explanation.html) revealed the presence of seven cysteine residues (C72, C196, C312, C337, C411, C432, and C495). Cysteines C312 and C337 and C411 and C432 were predicted to form disulfide bonds. However, the YdaP structure model predicted the distances between these cysteine residues to be approximately 3.5 Ǻ, compared to 2.0 Ǻ for typical disulfide bonds, suggesting that they do not form the disulfide bonds in vivo. It is known that disulfide bonds serve the critical function of stabilizing protein fold. Furthermore, it also plays a key role in oxidative, heat, and toxic element stresses (Leichert et al., 2003).
Catalytic Site Residues
The B. licheniformis YdaP showed considerable homology to the L. plantarum POX in the catalytic center [Figure 7]. Putative active site residues were identified in the YdaP model structure [Table 1], the putative active center was made up of a α-helix and two loops (Johnson et al., 1994; Jones, 1999), which were located at the subunit interface (Rapp and Friesner, 1999). In the model structure of B. licheniformis YdaP, the extended curved accessible channel was identified [Figure 7] and this channel was suggested to allow the accessibility of the substrate into the active site. The amino acid residues of this channel region were relatively less conserved (20%) in the structures of both YdaP (target sequence) and POX from L. plantarum (template sequence) (Wille et al., 2006). The residues Ile538, Ser539, Tyr540, Thr541, Val544, and Asn545 were potentially involved in the YdaP substrate binding site [Figure 8]. However, in L. plantarum POX these residues were located in different positions compared to B. licheniformis YdaP. There were considerable differences in the conservation of the substrate binding residues including Lys554, Leu555, Arg556, Leu557, Ala560, and Met561 [Figure 8], which were found in L. plantarum POX (Wille et al., 2006).
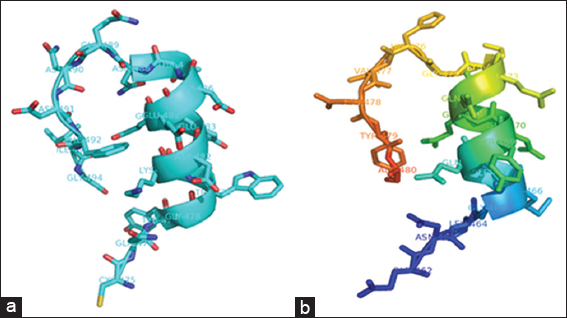
Figure 7: Representation of active center of (a) Lactobacillus plantarum pyruvate oxidase; (b) Bacillus licheniformis YdaP and residues thought to be involved in catalytic activity
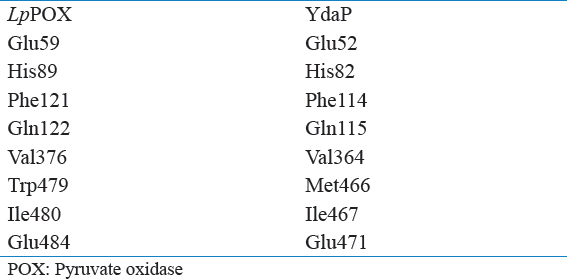
Table 1: Comparison of active site residues between Lactobacillus plantarum POX and Bacillus licheniformis YdaP
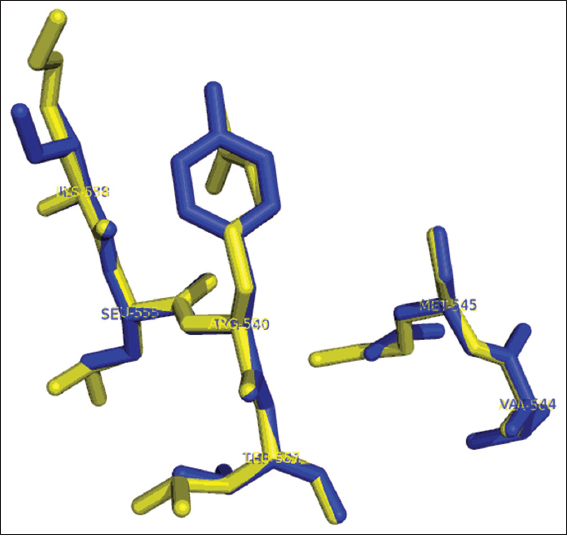
Figure 8: Stick representation of superimposition of residues involved in substrate binding site of Bacillus licheniformis YdaP (in blue) and Lactobacillus plantarum pyruvate oxidase (in yellow)
The 3D structural model of YdaP of B. licheniformis was constructed based on the closest similarity to the experimentally determined structure of LpPOX (Johnson et al., 1994; Wille et al., 2006). The structural model generated was assessed and it revealed to be in a good agreement with template structure suggesting the accuracy spectrum of the B. licheniformis YdaP model.
Active site residues were shown to be conserved among the two proteins except for Met466 of B. licheniformis YdaP which correspond to Trp479 in L. plantarum POX [Table 1] as well as in other homologues; however, the substrate binding residues have low conservation. The residues are clearly localized at the interface of the B. licheniformis YdaP subunit. Inspection of the active site residues revealed that Met466, Ile467, and Gln471 were located in the active site cavity. The active site residues were found to be similar in B. licheniformis YdaP compared to the L. plantarum POX [Table 1] which may possibly allow accessibility of other substrates. The classical features of B. licheniformis YdaP, include ThDP motif signature, catalytic active site, and disulfide bonds, were predicted and evaluated. The YdaP model structure suggests that the catalytic cavity comprised Met466, Ile467, Gln468, Gly469, Lys470, Gln471, Gln472, and Glu473 which were located on the α-helix of the active site cavity, while residues Lys474, Gly475, His476, Val477, Asn478, Tyr479, and Ala480 were present on the loop on the opposite site of the catalytic cavity. Overall comparison of the both structures of B. licheniformis YdaP and L. plantarum POX, beside the arrangement of the active center residues showed that there was some level of variation on residues conservation. However, the active center exhibited consistency on structural basis compared to the LpPOX template structure [Figure 7]. Interestingly, the model structure prediction did not provide adequate details of catalytic mechanism of this group of enzymes. Therefore, crystal structure data are required to elucidate on the catalytic mechanisms of YdaP protein. The presence of seven cysteine residues within the YdaP protein suggested that the B. licheniformis YdaP could form disulfide bonds. However, distance prediction of disulfide bonds by the B. licheniformis YdaP structure model was not appropriate for formation of the disulfide bonds (~2.0 Ǻ). Despite the utilization of the detergent (1% Triton X-100) for the YdaP enzyme activation, there was no role played by it as reducing agent. Therefore, it can be concluded that Triton X-100 did not affect the formation of the disulfide bonds (Lako et al., 2018).
CONCLUSION
The comparative modeling strategy could provide useful information for improving the characteristics of the YdaP protein, particularly in the identification of the active site residues [Table 1]. The YdaP model structure showed that Met466 Ile467 and Glu471 located in the catalytic cavity are believed to be involved in catalytic activity. However, Glu52, His82, Phe114, and Gln115 appeared to be located on the surface, played an important role in catalytic activity of B. licheniformis YdaP. These predictions provided some basic information that could be useful for future studies of particular residues which might be a potential target for site-directed mutagenesis studies for the improvement of the activity of the enzyme (Arnold, 1998).