INTRODUCTION
A germfree murine (GFM) animal is defined as a rat or mouse that has been maintained free from demonstrable microbial associates such as bacteria, viruses, fungi, and parasites throughout its life. Its sterile characteristics make it a unique animal model for exploring the relationship between the intestinal microbiota and the host. Using this model system, we can demonstrate the physiological and pathological effects of the intestinal microbiota on the host. Moreover, it is possible to investigate the roles of certain important microbes by manipulating GFM. The term gnotobiology is used to refer to studies of this kind. It is detected that the human microbiota possesses 1014 bacterial cells, a number 10 times greater than the total number of human cells (Bäckhed, 2009).
The first germfree (GF) animals were developed as far back as the 1800s by aseptic cesarean section, and the methodology used in the generation of GF mice remains unchanged mainly today. Evidence of grossly abnormal enteric plexus architecture and size in GF rats has been reported. Moreover, GF rats have been used to demonstrate the impact of the microbiota on migrating myoelectric complex activity. However, no change in ENS neurochemistry was observed following the colonization of these animals. More recently, the early postnatal developmental trajectory, neurochemical profile, and function of the ENS have been investigated in GF mice. In the context of my enteric nerve fiber density, a GF environment significantly decreased the development of the enteric neural network in a region-specific manner on postnatal day 3 relative to both offspring born in a specific pathogen-free (SPF) environment or to dams colonized with a simplified microbiota (Collins et al., 2014; Luczynski et al., 2016).
While our knowledge of the links between physiological functions and microbiota are growing constantly, notably thanks to the comparison between GF animals and conventionally raised conspecifics, recent studies indicate that the nature of the microbiota plays a role in the observed effects on animal physiology. One of the most striking examples comes from the studies of metabolic disorders. For example, transplantation into mice of fecal microbiota from human twins discordant for obesity induces different metabolic phenotypes (Ridaura et al., 2013). Similarly, transplantation of fecal microbiota from mice discordant for glucose metabolism induces different glycemia levels and glucose tolerance patterns in the recipient mice (Rabot et al., 2016; Le Roy et al., 2013).
Microbial communities are related to changes in gut morphology, physiology, and biochemistry. Microbes ferment polysaccharides and proteins, produce vitamins, and metabolize bile acids, affecting enterohepatic circulation and nutrient absorption. However, the implication of gut microbiota on host energy homeostasis remains elusive (Bäckhed et al., 2004), partly due to inappropriate analysis of energy expenditure data and spurious data interpretation. In obesity studies, normalization of energy expenditure for body size and composition variation requires proper statistical methods since lean mass is metabolically more active than fat mass, and simple body mass-specific ratios do not account for such differences. Hence, the influence of diet and host microbiome on energy balance must be analyzed carefully (Packard and Boardman, 1999).
In humans, the population of microorganisms is 10 times higher than host cells, which code a hundred times more genes than that of the human genes with 500–1000 different types and account for 30–50% of the total weight of feces. The gut microbes synthesize many essential vitamins for the development of the host immune system and hypothalamic-pituitary-adrenal (HPA) system and SE protein residues to formulate the non-essential amino acids and promote the absorption of mineral elements (Sudo et al., 2004). Recently, intestinal microbes’ protective and pro-inflammatory roles have gained increasing attention, as their disturbance is speculated to cause a significant number of diseases linked with metabolism and assimilation functions. In 2007, the Human Microbiome Project was initiated to expand our knowledge of the composition and functions of microbial communities in human health and disease. Previous metagenomic studies indicated that infections could change the composition and functions of the microbial communities, influencing the physiological features and gut homeostasis involved in the development of innate and adaptive immunity, intestinal immune tolerance, nutrients bioavailability, metabolism, energy harvest, and intestinal barrier function. However, it is still challenging to understand the influence and functional role of gut microbiota in shaping human health (Li et al., 2008) thoroughly.
In 1991, Dr. Margulis and Fester introduced the concept of the “holobiont.” This term is defined as the association of the host and its entire microbial community, including transient and stable members. The human microbiome is the assemblage of all microbiota that reside in tissues and biofluids, including the saliva, oral mucosa, skin, mammary glands, uterus, placenta, seminal fluid, ovarian follicles, airways, and gastrointestinal tract. It was often proposed that in the human body, bacteria outnumber human cells by a ratio of at least 10:1. However, in 2016, the ratio was revised and now it is estimated to be closer to 1:1. The vast majority of commensal bacteria reside in the gastrointestinal tract followed by the skin. The cecum and the colon are the dominant contributors to the total bacterial population within the gastrointestinal tract. As a result, the microbiota of the gastrointestinal tract and its interplay with systemic health are the most extensively documented.
So far, various animals have been employed as essential models to evaluate microbial functions or the effects of drugs or toxic materials on host health. Among those animal models, zebrafish and conventional mice own numerous advantages, such as having a small body, short lifespan, and high similarity to the human genome (Feitsma and Cuppen, 2008). Thus, those wild and transgenic zebrafish models have been applied to study human diseases and toxicology of environmental contaminants such as inorganic (e.g., heavy metals) and organic pollutants (e.g., endocrine disruptors), by evaluating the biochemical markers or endpoints related to acute toxicity, behavior, cell death, and transcriptional gene expression of signaling (Yang et al., 2009). Notably, the gut microbiota are highly sensitive to drugs, diet, and environmental pollutants, including persistent organic pollutants, antibiotics, heavy metals, pesticides, nanomaterials, and food additives (Jin et al., 2017). Thus, it is essential to evaluate the toxicity of chemicals and other stressors on gut microbiota and the subsequent effects on host health. At present, the zebrafish can be applied to investigate the mechanisms of pollutants toxicity by exploring the distribution, richness, and diversity of gut microbes, which are related to the accumulation of pollutants in the digestive tract, gut development, and health state, including the immunity, goblet cell density, and so on (Jia et al., 2019). However, the impacts of gut microbe on host health are not thoroughly investigated yet, partly due to the ambiguity in understanding the bacterial functions in vivo.
In recent years, GF or gnotobiotic animal models including mice, zebrafish, and swine are being employed as essential tools for studying the functional interactions of gut microbes in host health. Gut microbes assist in developing the mucosal barrier involved in food digestion and stimulate the immune system, which disruption can induce obesity, diabetes, cancer, and other human diseases (Wang and Donovan, 2015).
The in vivo gnotobiotic models also offer a more realistic environment to study microbe-microbe interactions than conventionally raised (CR) animals with the confounding intestinal microbiota (Martín et al., 2016). The GF animal models with a known set of microorganisms are increasingly applied in biomedical research by allowing researchers to control an animal’s microbiota precisely through the essential bacteria, genes, and diet factors. This strategy can be widely used to decipher host-microbe interactions in the development and function of the intestinal epithelium and the homeostasis effects on human health. Previously, GF mice and zebrafish larvae (5 days post-fertilization, dpf) have been successfully colonized with the human microbiota (Faith et al., 2010). However, due to various limitations in culturing, such as optimizing feeding pre-treatment and cleaning wastewater, the established GF zebrafish model could only be used for acute settings with early life stages.
Cancer is a disease initiated and progresses (through processes including tissue invasion and metastasis) through changes in the genome and epigenome. Hence, to establish a direct, causal connection between the gut microbiome and colorectal cancer (CRC) development, we must determine whether and how microbes alter mutation rates, gene methylation, chromatin structure, and/or non-coding RNA expression in CECs. Several epidemiological studies have associated specific bacteria in the gut with tumors characterized by DNA hypermethylation or specific mutational patterns (Burns et al., 2018), strengthening the hypothesis that gut microbes have a role in CRC development through their effects on the genome and epigenome of CECs. Gut microbes could elicit their effects on the genome or epigenome through direct or indirect mechanisms. There are two critical indirect mechanisms. The first is the capacity of gut microbes to induce a pro-carcinogenic inflammatory, and the second is the production of secondary metabolites by gut microbes response.
Bisulfite Sequencing of GF and Conventional Mice
Bisulfite genomic sequencing is regarded as a gold standard technology for the detection of DNA methylation because it provides a qualitative, quantitative, and efficient approach to identify 5-methylcytosine at single base-pair resolution. This method was first introduced by Frommer et al. It is based on the finding that the amination reactions of cytosine and 5-methylcytosine (5mC) proceed with very different consequences after the treatment of sodium bisulfite (Frommer et al., 1992). In this regard, cytosines in single-stranded DNA will be converted into uracil residues and recognized as thymine in subsequent PCR amplification and sequencing. However, 5 methylated cytosines (mCs) are immune to this conversion and remain as cytosines allowing 5 mCs to be distinguished from un-mCs. A subsequent PCR process is necessary to determine the methylation status in the loci of interest using specific methylation primers after the bisulfite treatment. The actual methylation status can be determined through direct PCR product sequencing (detection of average methylation status) or sub-cloning sequencing (detection of single molecules distribution of methylation patterns). Moreover, bisulfite sequencing analysis can identify DNA methylation status along the DNA single strand and detect the DNA methylation patterns of DNA double strands since the converted DNA strands are no longer self-complementary and the amplification products can be measured individually.
There are several advantages attributed to bisulfite sequencing in GF and conventional mice to reveal host-microbiota interactions. These include bisulfite-based DNA methylation analysis which has more quantitative accuracy, detection sensitivity, high efficiency, and a broad spectrum for sample analysis. As a fundamental method of DNA methylation analysis, bisulfite genomic sequencing has been widely used in various research and clinical settings. To optimize the final results of the bisulfite genomic sequencing protocol, numerous modifications have been explored and have significantly improved the sensitivity and accuracy in this procedure (Hajkova et al., 2002).
GF and Conventional Mice
Gnotobiotic mice (from the Greek gnotos for known and bios for life) refer to mice in which every microorganism present is defined. GF mice are one class of gnotobiotic animals, but mice associated with defined bacterial communities (e.g., altered Schaedler flora) are also considered gnotobiotic. Gnotobiotic animals are kept in isolators for long-term maintenance of their microbiological status, although various caging systems can allow for alternate husbandry solutions.
Conventional is an often used but poorly defined term in microbiome research. A conventional mouse is colonized with a diverse and largely undefined microbiome containing ~1011 bacteria within the gut alone. The exact microbial composition of a “conventional” mouse can vary widely, and there is no standardized “conventional microbiome.” The microbiome of a wild mouse differs widely from that of laboratory mice, which themselves can have very different microbiomes depending on how and where they are raised. A conventionalized mouse generally refers to a GF mouse that is colonized with the fecal or intestinal microbiome of a non-germ-free mouse.
Modern laboratory mice are often referred to as SPF, meaning that a defined list of mouse pathogens is excluded from these animal colonies. These specified pathogens vary between institutions, making SPF itself a vague term. Vendors often raise mice at defined health standards that provide various levels of exclusion for pathogens, opportunistic bacteria, and commensals. Apart from these excluded microorganisms, the exact microbiome of an SPF mouse is not generally known or controlled and is determined by the husbandry methods established for each health standard.
MATERIALS AND METHODS
Rearing of Model Animals
Male Albino mice (170–200 g) were purchased from Experimental Animal Market (Hong Kong Inc.) and were reared under controlled conditions (23 ± 2oC and 12 h/12 h light/dark cycle). The mice were fed with standard food and water ad libitum. Protocols were approved by the Laboratory Animal Care and Use Committee of Hong Kong National Pharmaceuticals by the National Institutes of Health Sciences guide for the care and use of laboratory animals.
Production of GF and Conventional Mice
Mice undergo a slightly different process due to lacking an egg life stage. To create a GF mouse, an embryo was created through in vitro fertilization and then transplanted into a GF mother. This process used a non-germ-free mother who was sacrificed and sterilized before the pups’ birth. After the cesarean birth, the pups were transferred to a sterile incubator with a GF mother for feeding and growth. These methods were only required for the generation of a GF mouse line. Once a line was generated, all progeny were GF unless contaminated. These progenies were then used for experimentation. Typically for experiments, each mouse was housed separately in a sterile isolator to prevent cross-contamination between mice. The mice were provided with sterilized food and water to prevent contamination. The sterilization methods varied between experiments because of different diets or drugs the mice were exposed. The isolators and waste products were continuously monitored for possible contamination to ensure complete sterility.
Revealing of Host-Microbiota Interactions
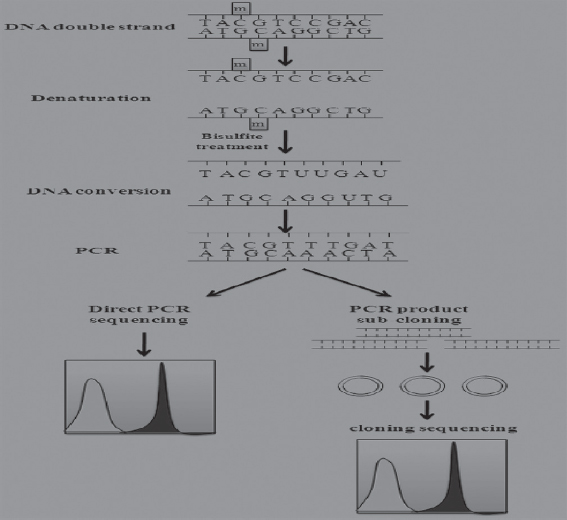
A sampling of intestines was performed on ice. For ileum samples, the distal 8 cm the small intestine was used, colon was sampled in whole. Both ileum and colon were rinsed with 0.9 % NaCl solution to remove intestinal contents and subsequently snap frozen in liquid nitrogen.
Whole-genome bisulfite sequencing
To prepare the library for bisulfite sequencing, we fragmented the genomic DNA of all the tissue samples to an average size of 100–300 bp through sonication (Covaris, Massachusetts, USA). The genomic fragments were end-repaired and TrueSeq methylated adaptors were ligated to their ends. Adaptor ligated genomic fragments were treated with sodium bisulfite as described in the previous study. Library preparation and sequencing were performed to generate 90-nt long reads in paired-end mode with sufficient sequencing depth (>30×) through HiSeq-2000 platform (Illumina, San Diego, USA).
Read alignment and identification of mCs
The adaptor sequences and low-quality reads were removed from the raw reads using NGSQC Toolkit (v2.3) at default parameters. Duplicated reads were filtered out by mapping on the rice genome (MSU v7.0) using Bismark (v0.8) under default parameters. The efficiency of bisulfite conversion was estimated by mapping high-quality filtered reads on rice chloroplast genome. More than 99% of the cytosines in the chloroplast genome were converted to thymine(s), indicating a very high efficiency of bisulfite conversion in our experiments. The mCs in mice genome were identified based on ≤0.001 P-value significance and sequencing depth of ≥5 reads, as described in the previous study (Garg et al., 2015). Methylation level was determined by estimating the percentage of reads giving methylation call at a particular cytosine site to all the reads covering that site in the sequencing data. DNA methylation patterns in mice genome were visualized through Circos plot using a Window size of 100 kb. The density of DNA methylation in genes/TEs and their 2 kb flanking regions was calculated using customized perl scripts.
RNA isolation and quantitative RT-PCR analysis
Total RNA was extracted from total tissue using the RNeasy Mini Kit (Qiagen). The purity and integrity of the RNA were assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies). For real-time PCR, 2 mg RNA was transcribed into cDNA using the reverse transcription system from Promega. Real-time quantitative RT-PCR analysis was performed using the Light Cycler LC 480 (Roche). Relative quantification was carried out using the LightCycler 480 SW 1.5.1 (Roche). GAPDH was used as a reference gene, relative mRNA expression values were calculated using delta-delta threshold cycle (Ct) analysis (Langmann et al., 2003).
Pharmacological Agents
Stock solutions of phenylephrine, acetylcholine, and sodium nitroprusside were prepared freshly in sterile and distilled water.
Statistical Analyses
The statistical procedures used included Student’s unpaired t-tests and one- or two-way analysis of variance (ANOVA). All analyses were performed using data analysis software GraphPad Prism 5.0 (USA, CA). The number (n) of the animals used is indicated in the graphs. Statistical significance was set at P < 0.05. The data are presented as mean ± standard error of the mean (SEM). For concentration-response curves, maximum response (efficacy or Emax) was calculated to show the highest point on the curve. Half maximal effective concentration (EC50) was calculated to evaluate the potency of the bisulfite sequencing.
RESULTS
Microbiota are Important for Vascular Mechanical Properties
Here, we observed for the 1st time that resistance arteries [Figure 1] from male GF mice present increased vascular stiffness, as shown by the leftward shift in the stress-strain curve and an increase in the elastic modulus b. Arterial stiffness is determined by the Young’s elastic modulus (E = stress/strain).

Figure 1: Rearing cage for mice under laboratory conditions
The stress-strain relationship is non-linear; therefore [Figure 2], it is essential to obtain a tangential or incremental elastic modulus (Einc) by determining the slope of the stress-strain curve. Taking derivatives on the equation presented in the method section, we see that Einc = βσ. For a given σ-value, Einc is directly proportional to β. An increase in β implies an increase in Einc, which means an increase in stiffness.

Figure 2: Germfree and conventional mice models
However, there was a distinct change in the type of remodeling between the sexes [Figure 3]. Arteries from male GF mice presented a decrease in cross-sectional area and luminal diameter, resulting in an inward hypotrophic remodeling characteristic for a chronic reduction in blood flow. On the other hand, arteries from female GF mice present an increase in cross-sectional area and luminal diameter resulting in an outward hypertrophic remodeling, a characteristic seen in aging.
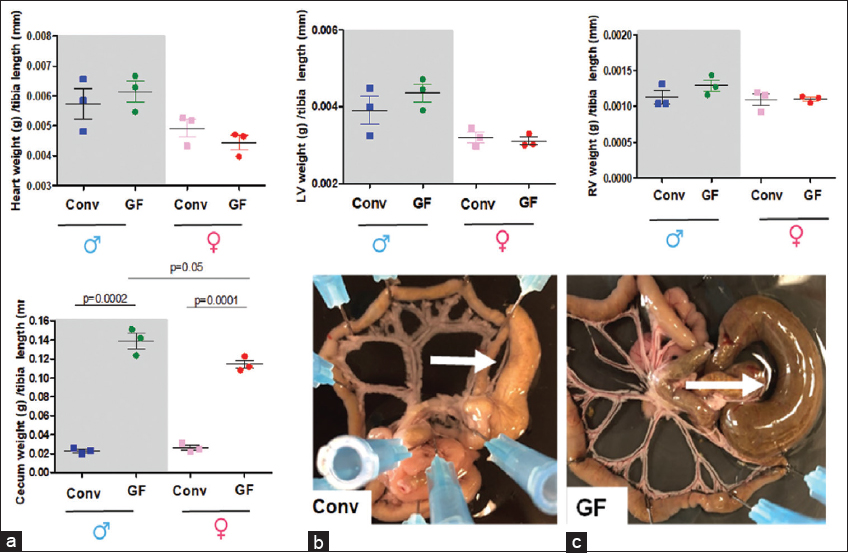
Figure 3: Bisulfite sequencing and vascular microbial properties in Conv and GF mice. Conv: Conventional, GF: Germfree
Total heart (A) left (LV, B) and right (RV, C) ventricles [Table 1] and cecum (D) weights were measured [Figure 4] and normalized by tibia length from 7 to 8 weeks old male and female Conv or GF mice. Number of animals used per group is = 3. Values are mean ± SEM. The images are representative pictures from the male Conv and GF mice (E). Arrows show the cecum. Please note that the mesenteric bed from the GF mice presents an enlargement of the cecum. This is a typical characteristic for GF animals.
Table 1: Emax to phenylephrine in resistance arteries of Conv and GF mice.
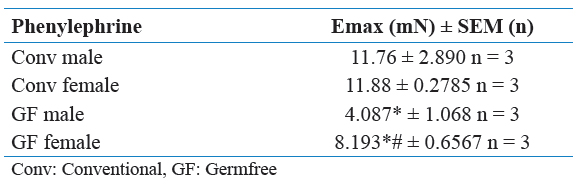
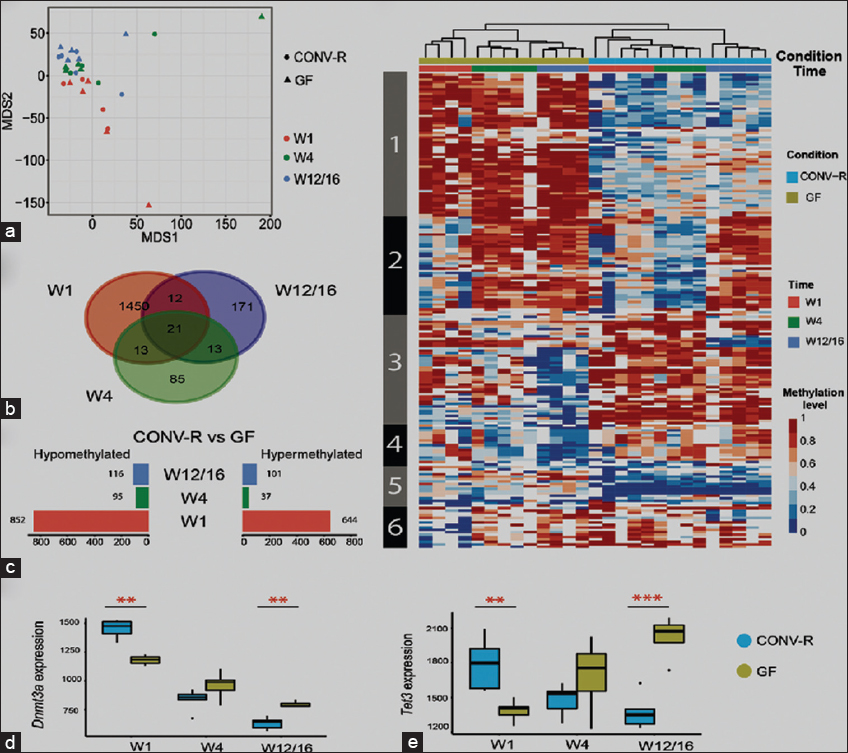
Figure 4: Postnatal development and the microbiota affect the DNA methylation profile. (a) Multidimensional scaling analysis plot displaying the overall methylation profiles. (b) Venn plots showing the number of differentially methylated sites between CONV-R and GF at the three developmental stages. Note the high number of differentially methylated sites at W1. (c) Number of hypo- and hypomethylated sites among all DMPs (CONV-R vs. GF) for each developmental stage. (d) Expression of Dnmt3a and Tet3 genes, which function in de novo methylation and demethylation, respectively. (e) Hierarchical clustering of differentially methylated sites between CONV-R and GF in the three developmental stages. Each row indicates a CpG site, and the color scale represents the methylation level. CONV: Conventional, GF: Germfree
Glycerophospholipid, Sphingolipid, and Gut-Microbiota Interactions
Lipids were quantified by electrospray ionization tandem mass spectrometry in positive ion mode as described previously. A precursor ion scan of m/z 184 specific for phosphocholine-containing lipids was used for PC (Liebisch et al., 2004). Neutral loss scans of 141 and 185 were used for PE and PS, respectively. Sphingosine (d18:1)-based ceramides were analyzed using m/z 264.
PE-based plasmalogens were analyzed according to the principles described by Zemski-Berry. Free cholesterol (FC) and cholesteryl ester (CE) were quantified using a fragment ion of m/z 369 after selective derivatization of FC using acetyl chloride (Liebisch et al., 2006). Correction of isotopic overlap of lipid species and data analysis by self-programmed Excel macros was performed for all lipid classes. For all lipid classes, non-naturally occurring lipid species were used as internal standards. Quantification was performed by standard addition calibration to tissue homogenates using several naturally occurring lipid species for each lipid class.
DISCUSSION
Over the past decade, our understanding of gut microbiota and its products in various diseases such as inflammatory bowel disease and metabolic disease has increased, and more recent reports have revealed the gut microbiota contributes to cardiovascular and renal diseases. In the contemporary era, given the rise of antibiotic-resistant microorganisms, increased number of immune compromised patients, and lack of new antimicrobial medications, researching pathogenic microbes remain highly relevant.
Furthermore, recent research strongly suggests that commensal [Table 2] gut microbiota colonizing the human body also substantially influence host physiology and the risk for common diseases. Indeed, our observations revealed that the commensal microbiota colonizing the mice body substantially affect vascular physiology in the present study. Specifically, our findings demonstrated that the interaction between host and microbiota, or the holobiont, is required for vascular homeostasis. Furthermore, our findings point to a holobiont-vascular network that is sex specific. These data provide new insight into how the holobiont and cardiovascular systems have crosstalk affecting vascular dynamics. Further, our results suggest that disturbances in microbiota synergy into the host are major risks for common diseases and may lead to new mechanistic discoveries.
Table 2: Bisulfite sequencing efficiency in terms of host-microbiota interactions
Commensal gut microbiota perform a variety of functions that are important to the host. Perhaps, one of the most apparent functions of gut microbiota is to help the host digest plant-derived complex carbohydrates and generates energy in short-chain fatty acids. Gut microbiota are also essential for the source of biotin, Vitamins K and B12, and essential amino acids. All these elements are essential for the well-being of the host. Therefore, based on these premises, we questioned whether the lack of microbiota would disrupt the host vascular homeostasis, specifically if it would induce changes in vascular function in young mice relative to Conv mice, and if these changes, if any, would be in a sex-specific manner.
CONCLUSION
The study of germfree and conventional mice thus insights to the host microbiota interactions.
Appreciation is growing for the role of non-dietary, environmental factors in obesity, including early-life events that impact intestinal microbes and regulate the host epigenome. However, specific microbiota-regulated targets that influence the obese phenotype are currently unknown. This can this be studied by comparing bisulphite sequencing of germfree and conventional mice. Inorder to elucidate the interactions among the microbiota, immune system, and epigenome in the context of obesity to facilitate future development of dietary strategies that modulate gut bacteria to prevent disease.
FUNDING
National Institutes of Health, United States.
REFERENCES
1. Bäckhed, F. 2009. Changes in intestinal microflora in obesity:Cause or consequence?
2. Bäckhed, F., Ding, H., Wang, T., Hooper, L. V., Koh, G. Y., Nagy, A., Semenkovich, C.F. and Gordon, J.I. 2004. The gut microbiota as an environmental factor that regulates fat storage.
3. Collins, J., Borojevic, R., Verdu, E.F., Huizinga, J.D. and Ratcliffe, E.M. 2014. Intestinal microbiota influence the early postnatal development of the enteric nervous system.
4. Faith, J.J., Rey, F.E., O'donnell, D., Karlsson, M., McNulty, N.P., Kallstrom, G. and Gordon, J.I. 2010. Creating and characterizing communities of human gut microbes in gnotobiotic mice.
5. Feitsma, H. and Cuppen, E. 2008. Zebrafish as a cancer model.
6. Frommer, M., McDonald, L.E., Millar, D.S., Collis, C.M., Watt, F., Grigg, G.W. and Paul, C.L. 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands.
7. Garg, R., Chevala, V.N., Shankar, R. and Jain, M. 2015. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response.
8. Hajkova, P., El-Maarri, O., Engemann, S., Oswald, J., Olek, A. and Walter, J. 2002. DNA-methylation analysis by the bisulfite-assisted genomic sequencing method. In:DNA Methylation Protocols. Springer, Totowa, New Jersey. pp. 143-154.
9. Jia, P.P., Sun, T., Junaid, M., Xiong, Y.H., Wang, Y.Q., Liu, L. and Pei, D.S. 2019. Chronic exposure to graphene oxide (GO) induced inflammation and differentially disturbed the intestinal microbiota in zebrafish.
10. Jin, Y., Wu, S., Zeng, Z. and Fu, Z. 2017. Effects of environmental pollutants on gut microbiota.
11. Langmann, T., Mauerer, R., Zahn, A., Moehle, C., Probst, M., Stremmel, W. and Schmitz, G. 2003. Real-time reverse transcription-PCR expression profiling of the complete human ATP-binding cassette transporter superfamily in various tissues.
12. Le Roy, T., Llopis, M., Lepage, P., Bruneau, A., Rabot, S., Bevilacqua, C. and Gérard, P. 2013. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice.
13. Li, M., Wang, B., Zhang, M., Rantalainen, M., Wang, S., Zhou, H., Zhang, Y., Shen, J., Pang, X., Zhang, M., Wei, H., Chen, Y., Lu, H., Zuo, J., Su, M., Qiu, Y., Jia, W., Xiao, C., Smith, L.M., Yang, S., Holmes, E., Tang, H., Zhao, G., Nicholson, J.K., Li, L. and Zhao, L. 2008. Symbiotic gut microbes modulate human metabolic phenotypes.
14. Liebisch, G., Lieser, B., Rathenberg, J., Drobnik, W. and Schmitz, G. 2004. High-throughput quantification of phosphatidylcholine and sphingomyelin by electrospray ionization tandem mass spectrometry coupled with isotope correction algorithm.
15. Luczynski, P., McVey Neufeld, K.A., Oriach, C.S., Clarke, G., Dinan, T.G. and Cryan, J.F. 2016. Growing up in a bubble:Using germfree animals to assess the influence of the gut microbiota on brain and behavior.
16. Margulis, L. and Fester, R., editors. 1991. Symbiosis as a Source of Evolutionary Innovation:Speciation and Morphogenesis. Mit Press, United States.
17. Martín, R., Bermúdez-Humarán, L.G. and Langella, P. 2016. Gnotobiotic rodents:An
18. Packard, G.C. and Boardman, T.J. 1999. The use of percentages and size-specific indices to normalize physiological data for variation in body size:Wasted time, wasted effort?
19. Rabot, S., Membrez, M., Blancher, F., Berger, B., Moine, D., Krause, L. and Chou, C.J. 2016. High fat diet drives obesity regardless the composition of gut microbiota in mice.
20. Ridaura, V.K., Faith, J.J., Rey, F.E., Cheng, J., Duncan, A.E., Kau, A.L. and Gordon, J.I. 2013. Gut microbiota from twins discordant for obesity modulate metabolism in mice.
21. Sudo, N., Chida, Y., Aiba, Y., Sonoda, J., Oyama, N., Yu, X.N., Kubo, C. and Koga, Y. 2004. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice.
22. Wang, M. and Donovan, S.M. 2015. Human microbiota-associated swine:Current progress and future opportunities.
23. Yang, L., Ho, N.Y., Alshut, R., Legradi, J., Weiss, C., Reischl, M. and Strähle, U. 2009. Zebrafish embryos as models for embryotoxic and teratological effects of chemicals.