INTRODUCTION
Alzheimer’s disease (AD) is a serious pathological disease defined through the accumulation of intracellular neurofibrillary tangles and extensive loss of neuronal and plaques by extracellular amyloid in the brain. According to clinical Phase, the sysmptoms is marked in AD by neurotransmitter loss, neuron inflammation, synaptic loss, and selective neuronal death (Holtzman et al., 2011). It was first described in 1906. Since the 1980s, AD was considered the serious cause of dementia in older adults. Therefore, in 1984, beta-amyloid protein was identified in the blood vessels of Down syndrome patients and AD suggested that the 21st chromosome presents like an extra copy in people with Down syndrome. AD was classified into three types according to their genetic change in the brain such as late-onset type, early-onset type, and familial type.
Apolipoprotein is a type of protein that binds lipids to form lipoproteins and transports the lipids in lymph, blood, and cerebrospinal fluid (Samy, 2014). There are several classes of apolipoprotein such as – A, B, C, D, E, F, H, L, and M, which play specific function for their region. The major risk factor is influencing a sporadic AD in the form of genotype for apolipoprotein E (apoE) (Blennow et al., 2006). The humans were showing polymorphism effect in the apoE gene with three different alleles (E2, E3, and E4), based on six different phenotypes (E2/2, E2/3, E2/4, E3/3, and E4/4) (Ashford et al., 2004).
The apoE was predominantly secreted through microglia and astrocytes (Pitas et al., 1987; Uchihara et al., 1995) in the brain while neurons should produce it (Aoki et al., 2003; Xu et al., 1999). The apoE is affects lipid-binding properties and three dimensional between isoforms (Mahley et al., 1996). The apoE is playing a key role in the uptake and transport of cholesterol with lipoprotein receptors, including the low-density lipoprotein (LDL) receptors at high-affinity interaction (Wilson et al., 1991).
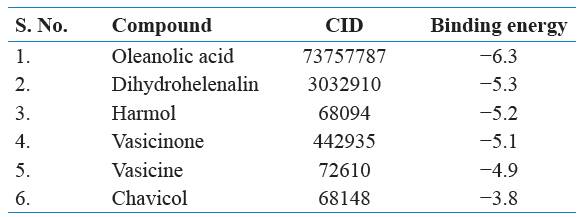
Molecular docking is used to screen the drug based on its structure and drug designing. The small molecules that interacted with the target protein were analyzed through docking. The molecular docking method is based on structural-based drug designing with ligand-binding sites in a protein of three-dimensional structures (Ferreira et al., 2015). According to computational approaches, docking helps with screening the compound sets which depend on free binding energies and hypotheses the molecules can inhibit their target (Abheepsa Mishra and Satyahari Dey, 2019). The comparison of docking molecules of proteins was an alternative to the drug-like properties from the initial molecule, which can allow to calculate their recorded values. In the present study, the natural compounds from different plant sources such as oleanolic acid, dihydrohelenalin (DH), harmol, vasicinone, vasicine, chavicol, and myrcene were selected against apoE for the treatment of ADs with the help of molecular docking.
MATERIALS AND METHODS
Identification of Protein by UniProt
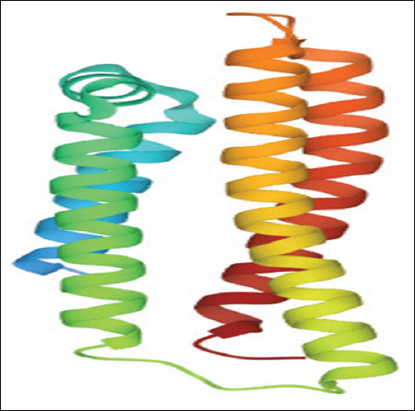
apoE is an associated protein with lipid particles and is mainly involved in lipoprotein-mediated lipid transport between organs through the interstitial fluids and plasma. It binds a cellular receptor including the LDL receptor and LDL receptor-related proteins have subclasses LRP1, LRP2, and LRP8 with low-density receptor/VLDVLR. They mediate the cellular uptake and transport of apoE containing lipoprotein particles. The structure of apoE (PDB ID 1GS9) was obtained by PDB https://www.rcsb.org/(Berman, 2000).
Retrieval of Ligands by PubChem
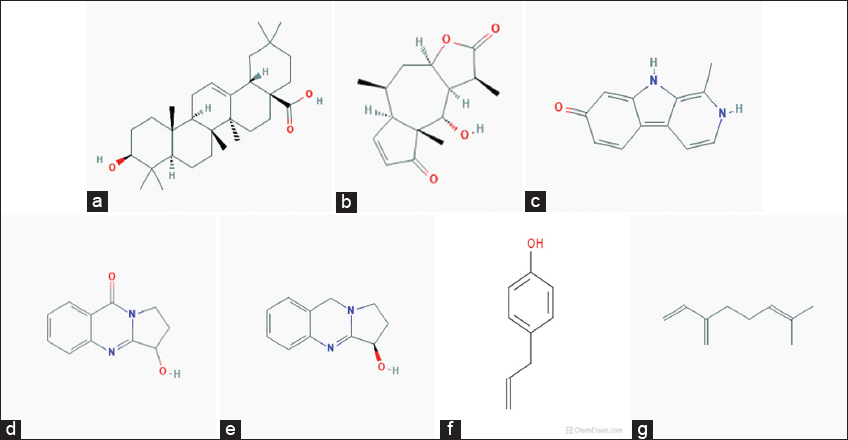
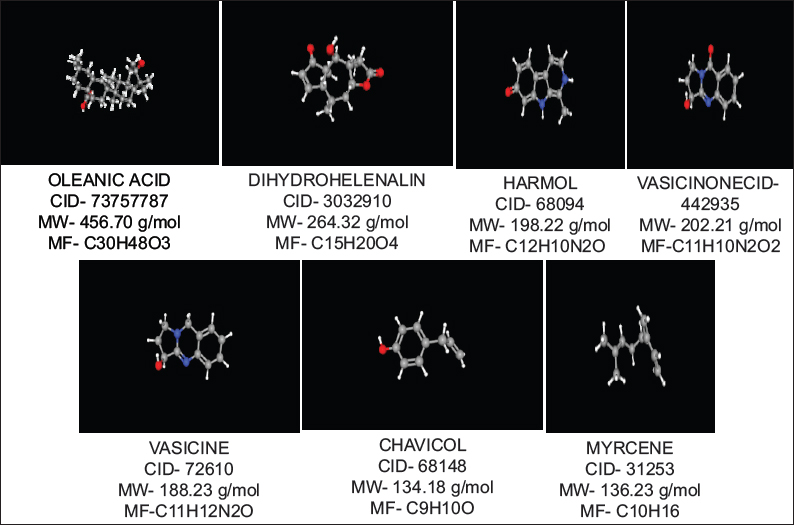
Oleanolic acid, DH, harmol, vasicinone, vasicine, chavicol, and myrcene were used for the study of docking from the literature. Oleanolic acid is a pentacyclic triterpenoid, commonly found in olive oil and also present in different plant species. It is inhibited of the cellular inflammatory process, known to induce Phase 2 xenobiotic biotransformation enzymes. The highest amounts were found in edible parts of jujube (Ziziphus jujuba Mill.), which is commonly consumed by fruit in South Asia and China (Guo et al., 2015). It is a derivative to induced both extrinsic and intrinsic apoptosis from multiple signaling pathways.
DH is found in Arnica Montana. The DH esters have low response factors against ultraviolet detection. DH is responsible for the activity of anti-inflammatory properties in Arnica montana L. (Asteraceae) and their extracts have short-chain esters of sesquiterpene lactones DH. DH is inhibited collagen-induced platelet aggregation, 5-hydroxytryptamine secretion, and thromboxane formation with a concentration-dependent manner at 3–300 mm. While DH is reduced, the number of acid-soluble sulfhydryl groups that is present in platelets increase up to 78% than their anti-aggregatory concentrations.
Harmol was a chemical compound, which classified as β-carboline alkaloids (Abe and Kokuba, 2013). It is manufactured by harmalol, harman, harmaline, and harmine, which are related to hallucinogenic alkaloids. Harmine was first identified from the seeds of Syrian rue (Peganum harmala), found in a wild desert shrub of Central Asia, Syria, and the Middle East. Other related alkaloids are derived from the bark and stems of ayahuasca (Banisteriopsis caapi). It was found in vivo in humans from O-demethylation of harmine (Morales-Garcia et al., 2017). The β-carboline alkaloids were naturally occurring plant substances, known as a wide spectrum of psychopharmacological, neuropharmacological, and antitumor effects.
Vasicinone is a form of quinazoline alkaloid that shows bronchodilatory activity in vitro and in vivo (Amin and Mehta, 1959; Mehta et al., 1963). It was showing anti-anaphylactic action which presents with Peganum harmala (Rajani et al., 2008). Vasicinone has used the leaf extracts of Adhatoda vasica Nees, was reported that vasicinone is the major metabolite of vasicine (Amin and Mehta, 1959). They show apoptotic properties in a cell-specific manner (Qazi et al., 2014) and inhibited the PI3K/Akt/FoxO3a pathways in vivo and in vitro.
Vasicine (peganine) is found in Justicia adhatoda and it’s a quinazoline alkaloid. In addition, vasicine is present in Peganum harmala (Moloudizargari et al., 2013). Their chemical formula is C11H12H2O and compared to theophylline both in vivo and in vitro (Nepali et al., 2012). They also have combination studies with their combination ratio (1:1) with bronchodilatory activity in vivo and in vitro. The alkaloids are used for respiratory stimulants (Avula et al., 2008). Vasicine is considered to have a uterine stimulant effect (Rajani et al., 2008).
Chavicol is a type of organic compound and a natural phenylpropene, which considers the benzene ring chemical structure with hydroxyl group and a propenyl group substitute. They have been found colorless liquid property with terpenes in betel oil. It is miscible with chloroform, alcohol, and ether. Their dimerization gives neolignan magnolol. It is used in odorant in perfumery and as a flavor that presents many essential oils including gardenia and anise (Lide, 2005).
Myrcene is also known as β-myrcene. Their chemical formula is C10H16 with 0.794 g/cm3 density. It is a type of alkene natural hydrocarbon, which may be classified due to monoterpene. Monoterpene is an isoprenoid precursor and is found in the composition of essential oil of different plants such as hops, bay, and cannabis (Behr and Johnen, 2009; Chyau et al., 1996). ᾳ-Myrcene consists of isomer 2-methyl-6-methylene-1, 7-octadiene, which not present in nature (Behr and Johnen, 2009). It is described from pyrolysis (400°C) of β-pinene, which is obtained by turpentine (Behr and Johnen, 2009) and directly from plants (Eggersdorfer, 2005).
All ligand molecules were selected by the natural compound of several plants. All these ligand molecules were retrieved in 3D structure in “.sdf” format from PubChem (https://pubchem.ncbi.nlm.nih.gov/) (Graff and Mansuy, 2008) and also. All ligands structures were downloaded and can convert into a “.pdb” file through an online server (https://cactus.nci.nih.gov/translate/) through SMILES Translator (Berman et al., 2000). The converting “.pdb” files were downloaded into “.pdb” format and run files into different tools and software’s (https://pubchem.ncbi.nlm.nih.gov/).
Protein Preparation
In protein preparation, the missing atoms in the incomplete residues are attached and the protein molecule is prepared by removing water molecules and active sites of extra ligands are also removed. The alternative conformations (disorder) are deleted along with modelling the missing loop regions in standardizing atom names (Kim et al., 2015). The docked molecule structure can be view at the time of software. This software was used for the protein molecule loaded in the graphical windows and under analyzed the view option of hierarchy. Those water molecules that were attached with ligand molecules are deleted through selecting atoms and the protein crystal structure will be saved in a.pdb format file. This.pdb format file of protein was used for docking.
Preparation of Grid Parameters and Virtual Screening by PYRX
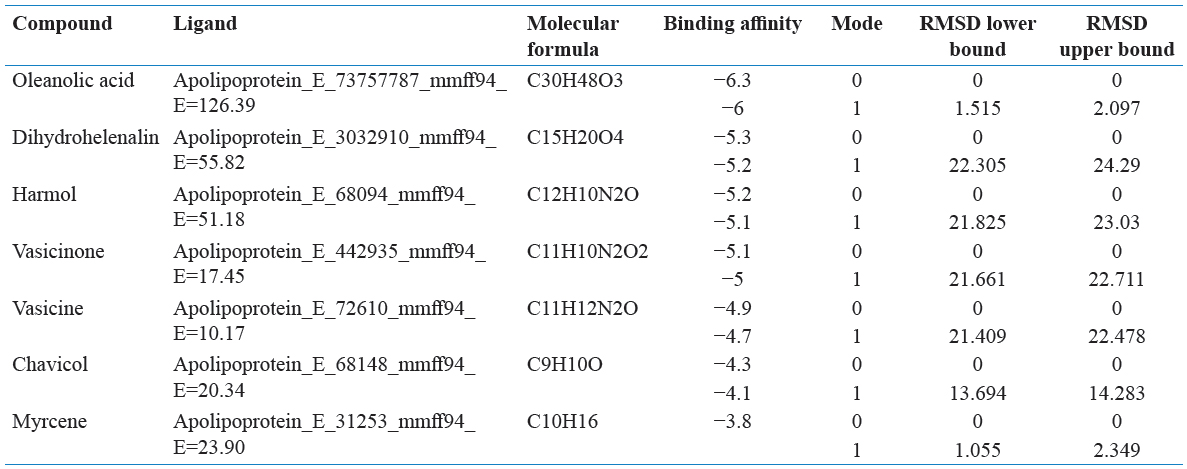
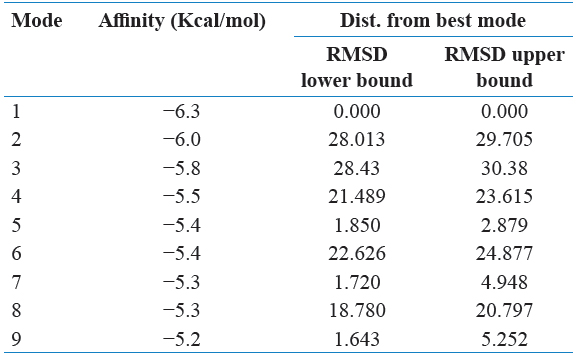
Grid maps were used to generate and spacing by 0.375 Å adjusted and cannot have ligand-binding properties. The grid dimension was adjusted to 40 × 40 × 40 points. AutoDock Vina was used to interacting with docking maps and values were calculated by AutoGrid. For each and every interaction between ligand atom and receptor, the entire binding energy of the site is calculated that represented in the grid. The selected protein of interaction energy was assigned at every one point of the grid and calculated their ligand affinity.
PyRx software was used for the future virtual screening of the ligands. This software was used to demonstrate the ligands with minimum binding energy with protein target through the virtual screening. Those ligands were found their minimum binding energy with a screening of drug likeliness property analysis. The protein molecule was loaded in the PyRx window and was converted the file from the.pdb format file to.pdbqt format file and then ligands were saved in.sdf format file folder. The minimum energy of ligands was converting the.sdf file to “.pdbqt” format. The results were analyzed based on their binding affinity.
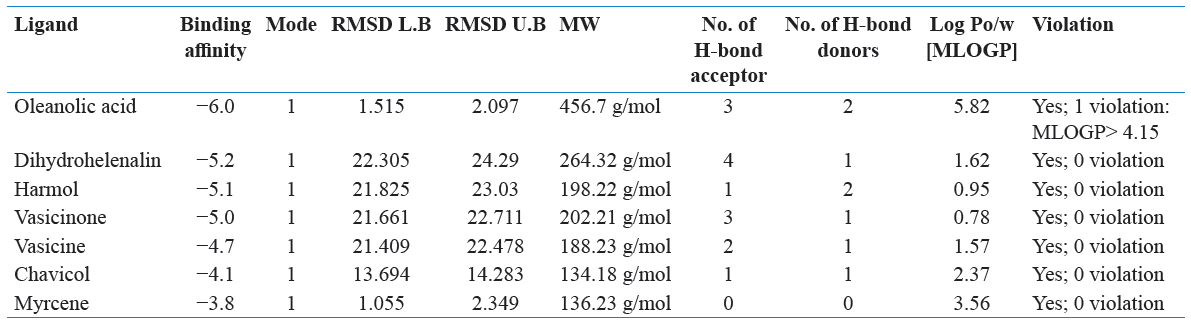
Drug Likeliness Property Analysis by SwissADME
Drug likeliness property was analyzed through an online server through SwissADME. The natural compounds were used for final molecular docking studies by screening those ligands which having drug-like properties. Canonical SMILE text ligands of notations were copied by PubChem and run on the SwissADME online web server (Wang et al., 2016). Ligands were selected by Lipinski’s rule of five. Lipinski rule of five has the following points:
-
Molecular mass is <500 Da
-
Hydrogen bond donors only less than 5 (<5)
-
High lipophilicity can be expressed as LogP <5
-
Hydrogen bond acceptors only allow less than 10 (< 10)
-
One violation value only considered.
The ligands were based on the Lipinski rule of five and used for final docking further through AutoDock Vina.
Molecular Docking Between Receptor Protein and Selected Compound by MGL Tools
The protein target file of.pdb format was loaded on AutoDock Vina graphical windows. The protein target file of.pdb format was prepared through docking by deleting of water molecules, adding hydrogen polar atoms, and adding Kollman charges of the protein molecule and protein file was further save in “.pdbqt” format file, and compounds were converting their.pdb format file into “. pdbqt” file format. Selected the grid box was docked region. Using Autodock result in command prompt and results were analyzed (https://cactus.nci.nih.gov/translate/.). Molecular docking was analyzed out between the target protein molecule and ligand.
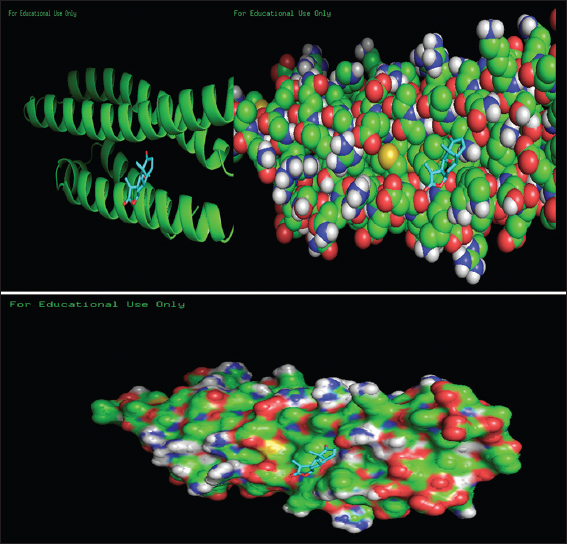
Structure Visualization Using PYMOL
Structure visualization was performed through the tool PyMOL 2.4. PyMOL 2.4 is a software and freely available tool in a website server. Selected the protein molecule from the “.pdbqt” format file and was uploading in their graphical showing screen of PyMOL 2.4 tool and imported the output “.pdbqt” file in the software. The docking structure of PyMOL 2.4 was visualized through the screen and may select the option molecule was converted into “molecular surface.” PyMol software was used for better visualization of protein and ligand interaction in the molecular surface. After AutoDock Vina, the output file was automatically saved in the selected folder with the name output.pdbqt file. This protein.pdbqt files with output.pdbqt files were loaded on their graphical screen of PyMol. The interaction between the protein and ligand was visualized and analyzed.